이전 글
https://novelism.tistory.com/259
다음 글
https://novelism.tistory.com/261
원하는 타겟 단백질에 대한 단백질 코드는 Uniprot 에서 확인하실 수 있습니다.


며칠 사이에 홈페이지 GUI가 업데이트 되었네요. 좌측은 예전, 우측은 바뀐 버전입니다.
검색창에서 TGFR1이라고 검색해봅시다.
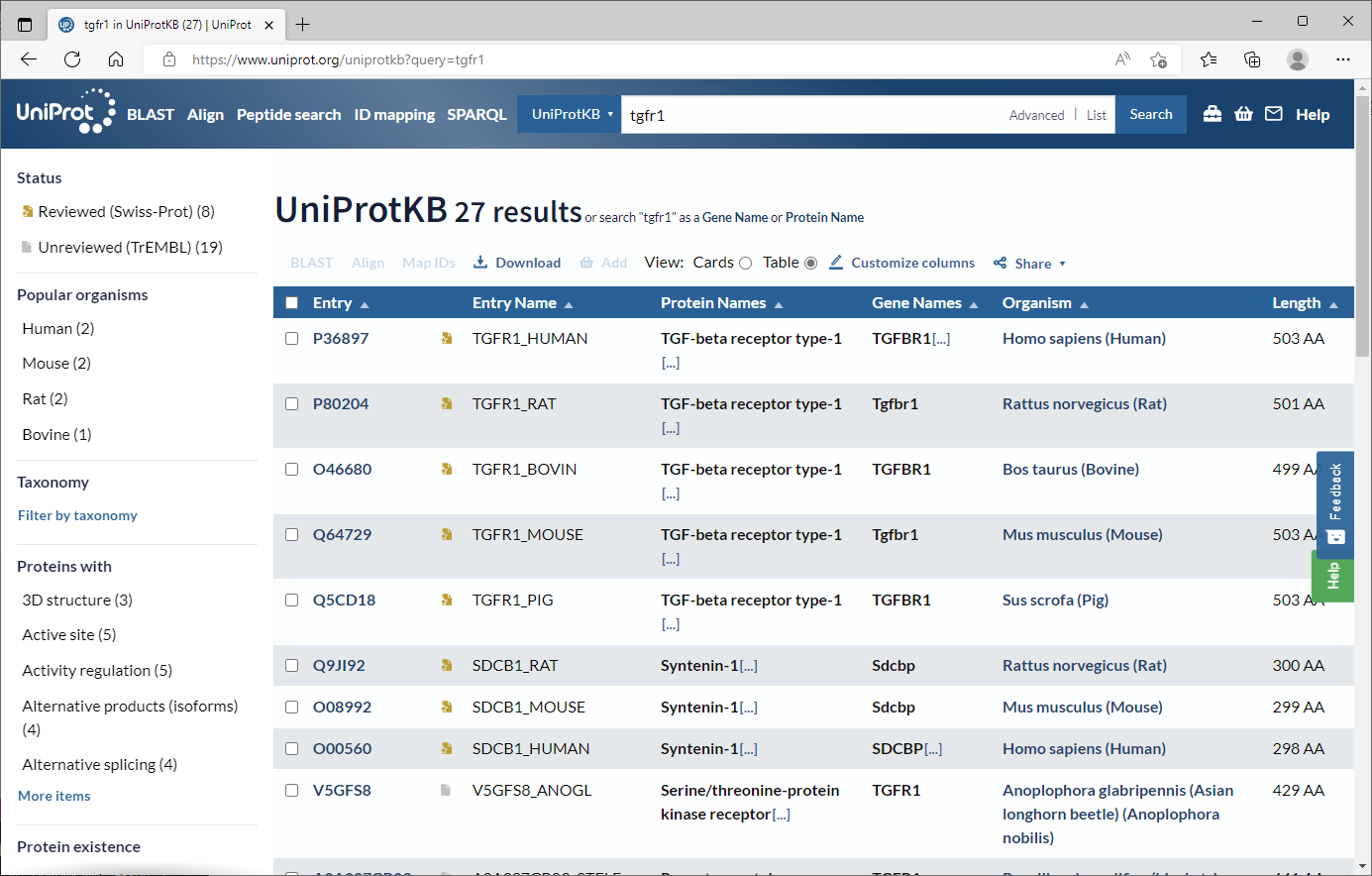
TGFR1_HUMAN에 해당하는 P36898을 클릭합니다.

쭈욱 내리다보면 Structure 가 나옵니다.
pdb 코드들은 여기서 얻을 수 있습니다.
저는 주로 이 리스트를 복사해다가 파일에 붙여 넣고 전부 pdb 구조를 다운로드한 후에 pymol 로 구조 정렬 후 살펴봅니다.
다음은 리스트의 pdb 들을 다운받는 코드입니다.
dw_pdb.py
#!/usr/bin/env python
import sys
import os
from urllib import request
usage = '''
dw_pdb.py list_file pdb_dir
'''
def main():
if len(sys.argv) < 2:
print(usage)
sys.exit()
list_file = sys.argv[1]
pdb_dir = 'pdb'
if len(sys.argv) >= 3:
pdb_dir = sys.argv[2]
fp = open(list_file)
lines = fp.readlines()
fp.close()
if not os.path.exists(pdb_dir):
os.makedirs(pdb_dir)
for line in lines:
lis = line.strip().split(';')
pdb_id = lis[0]
# method = lis[1]
# resolution = lis[2]
# chain_positions = lis[3]
print(pdb_id)
line_pdb = 'https://files.rcsb.org/download/%s.pdb' % pdb_id
pdb_file = '%s/%s.pdb' % (pdb_dir, pdb_id)
request.urlretrieve(line_pdb, pdb_file)
line_fasta = 'https://www.rcsb.org/fasta/entry/%s' % pdb_id
fasta_file = '%s/%s.fasta' % (pdb_dir, pdb_id)
request.urlretrieve(line_fasta, fasta_file)
if __name__ == "__main__":
main()
실행은
anaconda prompt에서
python dw_pdb.py list_file dir 입니다.
라고 치면 됩니다. 실행할 폴더에 dw_pdb.py 파일이 있어야 합니다.
list.txt 파일에 있는 목록을 pdb 라는 폴더에 다운로드하고 싶다면
python dw_pdb.py list.txt pdb
라고 입력하면 됩니다.
list.txt 파일의 예시는
1B6C;X-ray;2.60 A;B/D/F/H=162-503
1IAS;X-ray;2.90 A;A/B/C/D/E=162-503
1PY5;X-ray;2.30 A;A=175-500
같은 형식이나
1B6C
1IAS
1IAS
1PY5
처럼 ID 만 있어도 됩니다.
anaconda prompt에서 pymol 을 치면 pymol 이 실행됩니다.
pymol GUI 창에 다운로드한 pdb 파일들을 드래그해서 넣으면 입력됩니다.
pymol 창에서 alignto 3HMM을 입력하면 구조 정렬이 진행됩니다.

구조 정렬 진행 후, 화면을 우클릭하면 다음 같은 창이 나옵니다.
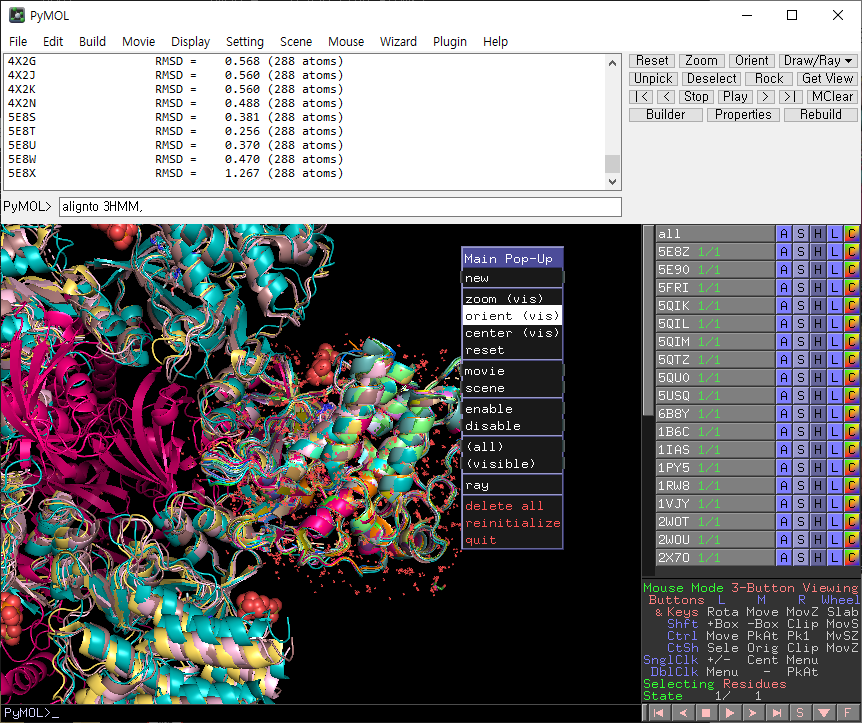
orient를 클릭하면 보기 쉽게 화면이 회전합니다.
pymol에선 마우스 우클릭 후 마우스를 움직이면 이 줌인 줌아웃
좌클릭 후 마우스를 움직이면 회전
휠을 클릭한후 움직이면 평행 이동
휠을 돌리면 초점거리(?) 변경입니다.
적당히 회전시켜서 리간드가 붙은 위치를 확인합니다.


pdb 코드 이름을 클릭하면 해당 pdb를 표시/비표시를 변경할 수 있습니다. all을 누르면 전부 표시/비 표시됩니다.
all을 한번 클릭한 후, 3HMM을 클릭합니다.
3HMM 옆의 A를 누르고, preset ligand sites cartoon 을 선택합니다.
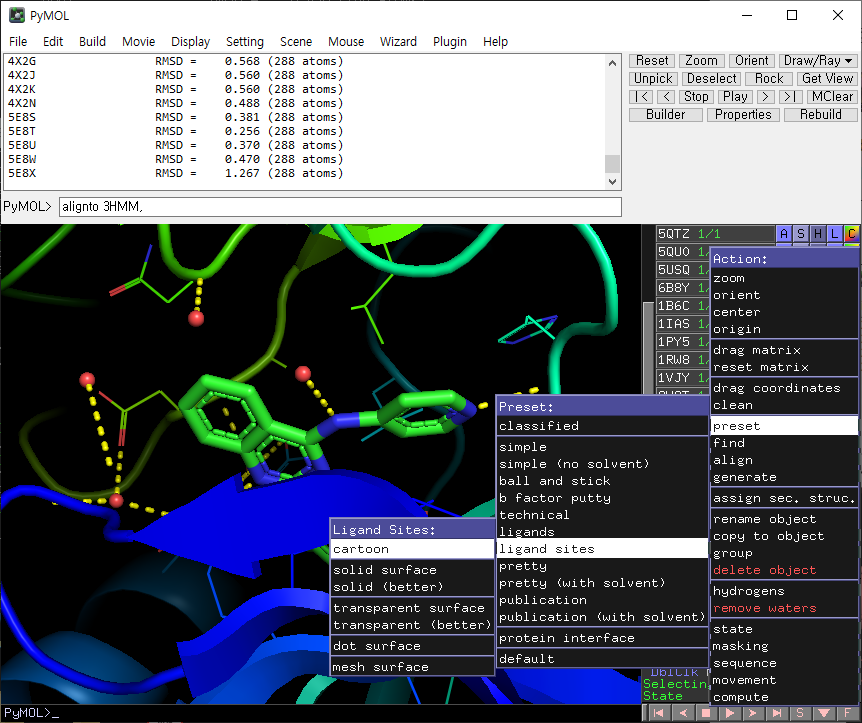
이렇게 하면 단백질과 리간드의 상호작용을 볼 수 있습니다.
상호작용 분석은 이전 글
https://novelism.tistory.com/253
https://novelism.tistory.com/256
글을 참고하세요.
3HMM이외에도 다른 pdb의 ligand들을 확인하면서 docking에 사용할만한 pdb 구조를 선택합니다.
docking이 잘되는 pdb 구조를 정하는 것이 좋은데 ( 크로스 도킹이 잘되거나, 알려진 데이터셋에 대한 실험값과 docking값의 코릴레이션이 높은),
보통 테스트를 해보기 전에는 그 기준으로 가지고 있는 리간드와 유사한 리간드가 붙어있는 구조, 정확도가 높은 구조 (missing atom이 적은), 등을 우선적으로 사용합니다.
단백질-리간드 결합 시 induced fit이 일어나는데, unbound form이나 ATP bound form보다는 이미 inhibitor가 붙어있는 구조가 아무래도 더 적절한 경우가 많습니다. 사실 여기에 만능의 정답은 없고... 테스트를 많이 해보고 그중 적합한 것을 선택하는 것이 제일 좋습니다. 필요한 경우 시뮬레이션으로 구조를 변경할 수도 있습니다.
이 경우에 저는 3HMM을 선택하였습니다. 사실 별 이유 아니고, DUD-E에서 그 pdb 구조를 사용했기 때문입니다.
'Drug > Computer-Aided Drug Discovery' 카테고리의 다른 글
| 알파폴드가 예측한 단백질 구조는 완벽한가? (3) | 2022.08.05 |
|---|---|
| chimera GUI에서 docking (vina) 사용법: 3. chimera로 도킹하기 (0) | 2022.07.03 |
| chimera GUI에서 docking (vina) 사용법: 1. 프로그램 설치 (0) | 2022.07.03 |
| conda 로 pymol, openbabel 설치시 주의사항 (0) | 2022.06.26 |
| TGFR1 DUD-E dataset 단백질 구조기반 분자 선별 (3) | 2022.06.20 |