백질 구조 기반 약물 가상 탐색: 1. 서론
https://novelism.tistory.com/116
서론에 이어서 단백질 구조 및 Cheminformatics로 ACE inhibitor (억제제)를 탐색하는 예시를 보여드리겠습니다.
고혈압 치료제로 사용하는 ACE억제제에 대한 포스팅은 이전 글을 참고하세요.
https://novelism.tistory.com/108
제가 구조기반 약물 탐색에서 하는 일은 3가지입니다.
- 단백질-약물 결합에서 중요한 결합기를 찾고, 해당 결합기를 가지는 약물들을 탐색합니다.
- docking을 통해서 약이 결합 위치에 들어갈 수 있는지 시뮬레이션하고,
- 중요하다고 생각한 결합기가 docking 결과 실제 예상한 위치에 있는지 확인합니다.
일단 이것은 예제이기에 분석과 분자 탐색은 적당한 수준까지만 진행하였습니다. 탐색된 분자에 대해선 책임지지 않겠습니다.
ACE 대해서 구조기반 접근 방법을 어떻게 사용하는지 보여드리도록 하겠습니다.
예제로 ACE를 선택한 이유는 ACE가 연구가 잘 되어있고, 구조도 잘 알려져 있고 기존에 개발된 약물이 많기 때문입니다.
혹시나 찾은 약물을 여기에 올렸는데 그게 좋은 약물이면 곤란하니까요. 그리고 DUD-E dataset에도 ACE가 포함되어있습니다.
ACE는 펩타이드를 가수 분해하는 기능을 한다고 이전 포스팅에서 이야기했습니다.
10-mer -> 8-mer + 2-mer
이 기능을 에서 중요한 부분은 ZINC와 cation을 가지는 residue입니다. 단백질 구조 기반 접근을 하려면 단백질 구조를 알아야 합니다. 여기선 펩타이드가 결합되는 포켓을 탐색하여 펩타이드에 경쟁적으로 단백질에 결합하여 펩타이드의 결합을 방해하는 약물을 탐색하도록 하겠습니다. 단백질 구조에서 포켓이 어떻게 생겼는지 확인을 해봐야 합니다.
uniprot에서 ACE HUMAN을 찾아봅니다.
https://www.uniprot.org/uniprot/P12821
ACE - Angiotensin-converting enzyme precursor - Homo sapiens (Human) - ACE gene & protein
Angiotensin-converting enzyme ACE Homo sapiens (Human) Reviewed-Annotation score: Annotation score:5 out of 5 The annotation score provides a heuristic measure of the annotation content of a UniProtKB entry or proteome. This score cannot be used as a measu
www.uniprot.org
서열이 1300이상으로, 640 정도를 기준으로 두 개의 영역으로 나뉘는 것을 볼 수 있습니다. pdb 구조들도 그중 한 영역만을 담고 있습니다. (알파폴드는 시뮬레이션이니까 제외합니다.) 타깃으로 하는 포켓은 C-terminal 도메인에 속해있습니다.
여기서 해당되는 도메인에 대 단백질 구조들을 찾아봅시다.
C-terminal을 포함하는 PDB ID는 다음 목록의 25개입니다.
1O86 1O8A 1UZE 1UZF 2IUL 2IUX 2OC2 2XY9 2YDM 3BKK 3BKL 3L3N 4APH 4APJ 4BZR 4C2N 4C2O 4C2P 4C2Q 4C2R 4CA5 6F9T 6F9U 6H5W 6ZPU
이것들을 다운로드해봅시다.
#!/bin/bash
mkdir pdb
line_pdb="wget"
for code in 1O86 1O8A 1UZE 1UZF 2IUL 2IUX 2OC2 2XY9 2YDM 3BKK 3BKL 3L3N 4APH 4APJ 4BZR 4C2N 4C2O 4C2P 4C2Q 4C2R 4CA5 6F9T 6F9U 6H5W 6ZPU
do
line_pdb+=" https://files.rcsb.org/download/$code.pdb"
done
echo $line_pdb
$line_pdb -P pdbpymol에서 pdb 파일들을 전부 읽어봅시다.
pymol pdb/*.pdbpymol 내에서 구조들을 정렬하고 싶다면, pymol의 명령 프롬프트에
alignto 1O86라고 입력하면 됩니다.
1O86에 포함된 리간드는 lisinopril입니다. enalaprilate (pdb_id: 1 UZE 참고)와 유사한 구조를 가지고 있습니다.
pymol 화면 우측 창에서 all을 눌러서 전체 구조 선택을 해제하고 1O86만 선택해봅니다. 그리고 1O86 옆의 A 버튼( Action)을 누르면 preset 이 있는데, 여기서 ligand sites-> cartoon을 선택합니다. 그러면 아래 같은 그림이 나옵니다. (금속이온을 좀 이상하게 표현해서 마음이 안 들지만) 단백질과 리간드의 상호작용을 살펴보기 좋습니다.
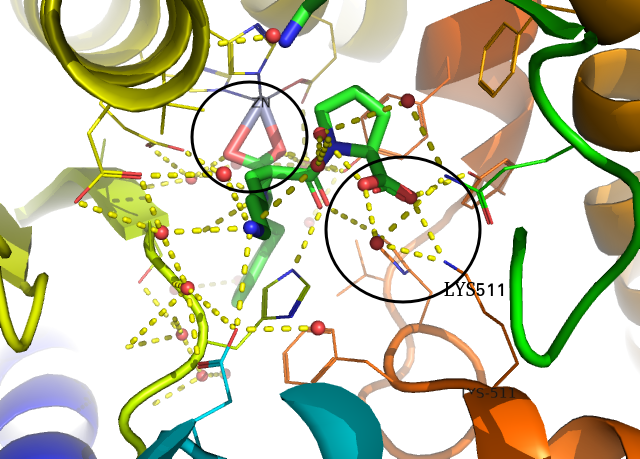
여기서 특별히 중요한 것이 ZN과 LYS511입니다. ZN2+나 LYS나 +전하를 가지고 있고, 이들에 대해 각각 리간드의 카복실기가 상보적으로 결합하고 있습니다. 또한 LYS511 근방에서는 water가 수소결합을 매개하고 있는 것처럼 보입니다.
만약 새로운 약물을 디자인한다면, 이 두 부분은 건드리지 않고, 다른 부분만을 고치는 것이 쉬운 선택지일 것입니다.
다른 약물을 살펴봅시다. PDB ID: 3BKL 입니다.

스케폴드는 다르지만, Zn2+, LYS511과 상호작용하는 부분은 위와 유사합니다. ZN 과 상호작용하는 부분이 카복실기는 아니고, 수소가 떨어져 나가서 형식 전하 (formal charge)가 음전하가 되기는 어려울 수도 있지만, 부분적으론 음전하를 띌 것으로 보입니다.
1UZF (ligand: captopril)처럼 Zn과 결합하는 부분이 SH (티올기)인 경우도 있지만 , 이 약물은 이미 부작용이 알려져 있고 이를 해결하기 위해서 티올기가 없는 ACE 억제제들이 개발되었으니 티올기를 가지는 구조들은 일단 배제하겠습니다. 탄소가 인으로 바뀐 경우도 있고, 4BZR 처럼 아예 다른 분자들과는 다르게 LYS511과 결합하지 않는, 포켓에 대한 결합구조가 상당히 다른 분자도 있지만, 대부분은 Zn2+, LYS511과의 결합을 가지고 있습니다.
아쉽게도 Fimasartan과 Valsartan의 3차원 결합구조는 안 보이네요. 약물 구조로 보면 두 약물은 같은 계열 같은데, Valsartan에는 카복실기가 하나 있지만, 이게 어디에 결합하는지는 잘 모르겠습니다. 시뮬레이션을 좀 해보면 적당히 알 수 있을 수도 있고요.
일단은 PDB에 있는 약물에 대해서만 구조를 확인해봤습니다. 보통은 ChEMBL에 있는 분자도 다 찾아서 비교를 해보는데, 굳이 거기까지 하고 싶진 않네요.
이 구조들을 보고 약물 탐색 조건을 만든다면, +charge를 가지는 LYS511과 상호작용할 수 있는 -charge를 가지는 결합기 하나 (예시: 카복실기), ZN2+와 결합할 수 있는 결합기 (예시: 카복실기) 하나를 가지는 것을 제일 중요하게 생각할 것입니다.
2개의 결합기를 가지면서, 두 결합기의 사이가 적당한 거리만큼 떨어져 있는 (위의 그림에서 분자를 그래프로 취급한다면, node 사이의 distance가 6.) 분자들을 검색한 후, docking 시뮬레이션을 합니다. 두 결합기가 위의 레퍼런스 구조와 유사한 위치에 있는지 확인한다면, 아마도 포켓에 결합할 가능성이 높은 분자들을 얻을 수 있을 것입니다.
비어있는 포켓에 대한 적합성이 높은 것들이 docking score가 잘 나올 것이라 믿고 다른 포켓에 대한 룰은 추가하지 않겠습니다.
Zn2+ 와 결합할 수 있는 결합기는 더 여러 종류가 있긴 하지만, 그건 그거대로 큰일이라서 굳이 카복실기 이외에 대해서 탐색하진 않겠습니다. 하지만 실제로 탐색을 진행하다보면 조건을 만족하는 분자가 너무 많거나, 딱 봐도 영 아닌 분자들이 많아서 룰을 더 세세하기 만들기도 합니다.
이렇게 룰과 도킹을 함께 사용하면, docking 만을 사용하는 것보다 선별된 분자의 유효성이 더 높을 것으로 기대할 수 있지만, 바이어스 문제가 있어서 분자의 신규성이나 다양성이 부족해질 수도 있습니다. 또한 룰에서 벗어난 분자는 설령 그것이 실제로 유효한 분자일지라도 유효한 분자로 예측되지 못한다는 단점이 있습니다.
'Drug > Computer-Aided Drug Discovery' 카테고리의 다른 글
| Protein-ligand binding affinity 예측에 대해서 (0) | 2021.10.25 |
|---|---|
| openbabel 사용법: obabel (0) | 2021.10.25 |
| 단백질 구조 기반 약물 가상 탐색: 1. 서론 (0) | 2021.10.14 |
| rdock: protein ligand docking 프로그램 (2) | 2021.10.05 |
| 분자 SMARTS 문법 및 rdkit에서 SMARTS 탐색 (0) | 2021.09.23 |