머신러닝 결과를 해석하는 것은 쉬운 일은 아니지만, 불가능하진 않습니다.
기본적으로 머신러닝은 통계분석의 응용이기에 주어진 데이터를 학습하고, 새로운 데이터를 예측할 때, 학습 데이터 중에서 주어진 데이터와 유사한 것이 있다면 그 영향을 크게 받습니다.
하지만 유사성이라는 것은 그리 간단한 것이 아닙니다. 입력 공간상의 거리(distance)와 임베딩 된 공간상에서의 거리는 다릅니다. 적절한 공간 변환을 일으키고, 유사성을 최적화하는 것이 딥러닝의 핵심입니다.
즉, 입력 공간에서 별로 유사해 보이지 않는 데이터가 임베딩 된 공간에선 한 곳에 모을 수 있습니다.
이것이 왜 중요한지 생각해봅시다.
우리는 굳이 딥러닝을 사용하지 않더라도, 분자의 fingerprint를 만들고, Tanimoto 같은 similarity를 정의하면 이를 이용해서 유사 분자들을 탐색할 수 있습니다. 그런데, 이렇게 탐색된 분자는 주로 스케폴드 구조적으로 유사합니다. 유효물질을 탐색한 후에 analog 탐색하기엔 적절하지만, 신규물질을 탐색하고 싶을 땐 특허를 피할 만큼 새로운 분자를 탐색하기엔 적합하지 않습니다. 뭐... 적당히 유사성을 낮추면 나오긴 하죠. 하지만 탐색되는 분자가 너무 많아집니다.
즉, 딥러닝을 통해서 우리가 기대하는 것은, 비슷하긴 한데, 특허는 피할 수 있는 분자입니다. 사실 기존 신약개발에서도 이미 많이 사용하는 전략입니다.
단백질과 약물의 상호작용 관점에서 생각해봅시다. 예를 들어 5-membered aromatic ring과 6-membered aromatic ring 이 있다면, 두 링이 aromatic ring이라는 점에선 유사하게 작용할 수 있습니다. 혹은 바이오 등전 자족 같은 경우, 뼈대 구조는 다르지만 3D 공간에서의 전자구조나 단백질-약물 상호작용에서의 역할은 유사하다고 기대할 수 있습니다.
즉, 뼈대 구조의 유사성을 학습하는 것이 아닌, 3D 공간 상호작용에서의 유사성을 학습할 수 있다면 training set에 있는 분자와 유사한 분자에 대해서만 예측하는 것이 아니라, 좀 더 다양한 분자에 대한 예측이 가능해질 것으로 기대할 수 있습니다.
A Deep Learning Approach to Antibiotic Discovery라는 논문을 예로 들어봅시다.
(Stokes, Jonathan M., et al. "A deep learning approach to antibiotic discovery." Cell 180.4 (2020): 688-702.)
이 논문에선 머신러닝을 활용해서 베타-락탐 계열 항생제가 아닌, 새로운 계열의 항생제를 찾으려는 연구에 대해 보고하였습니다. 페니실린으로 대표되는 베타-락탐 계열 항생제는 많이 사용되고 있긴 하지만, 내성이 문제가 되고 있다는 사실은 이미 유명합니다.
이 논문에선 딥러닝을 이용해서 베타 락탐 계열이 아닌 SU3327이라는 물질을 발견하였고, 실험 결과 항생제로서 약효를 확인하고 halicin이라는 새로운 이름을 붙여주었습니다. 이 물질을 찾은 딥러닝 방법은 directed MPNN (D-MPNN)입니다. 흥미로운 점은, Morgan fingerprints를 사용한 메서드(feed forward DNN, SVM, RF)들과 비교하였는데, 이런 방법들로는 halicin이 높은 점수를 얻지 못했다는 것입니다.

그러면 대체 d-MPNN은 어떻게 이 물질을 탐색했는가 라는 의문이 생깁니다.
그래서 training set의 active물질들 중 halicin과 유사하게 생긴 물질이 없었는지 확인해봤습니다.
의외로 많았습니다. active 분자가 110인데, 그냥 찾다 보니 38개만 봐도 그중에 9개 정도 있습니다. 다음은 그 목록들입니다.



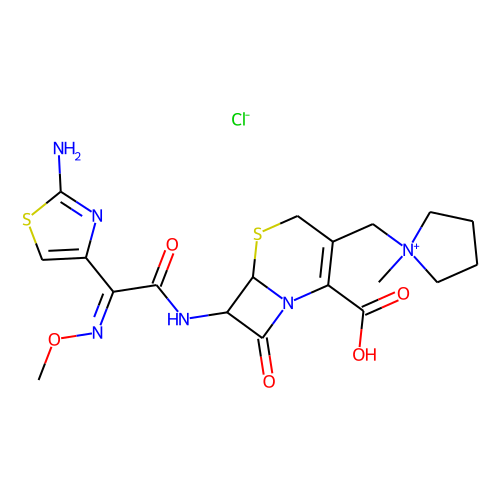


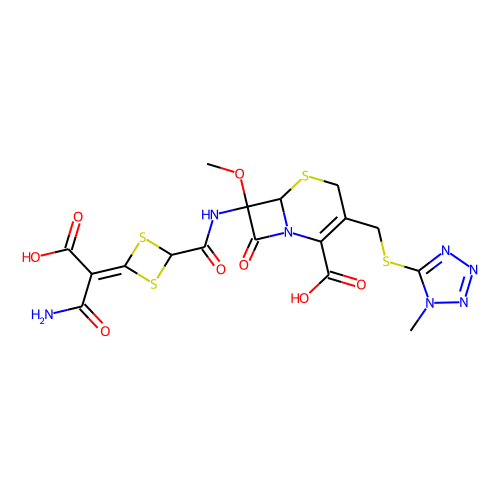


halicin을 예측하는데 영향을 미쳤을 것으로 추정되는 이 물질들은 대부분 황과 질소를 포함한 5-membered-ring을 가지고 있습니다. 하지만, halicin과 전체적인 유사성은 높지 않습니다. 아무튼 halicin이 적어도 부분적으로라도 학습 셋에 유사한 구조가 있었기에 예측된 것이지, 전혀 무관하게 튀어나온 것은 아니라고 생각할 수 있습니다. 그런데 왜 Fingerprint를 사용한 방법들에선 예측 결과가 다른 것일까요?
그런데, 재미있게도 이 물질들은 베타-락탐 구조를 가지고 있습니다. 즉, 이 물질들의 작동 기전은 전부 일치할 가능성이 높습니다. 그런데, halicin은 베타-락탐 구조를 가지고 있지 않고, 약리 기전도 다르다고 합니다. 타깃 단백질이 다른데, 약효는 있습니다. 좀 놀라운 일입니다. 아마도 얻어걸린 게 아닐까 좀 조심스럽게 추측합니다.
설령 이것이 얻어걸린 것이라 할지라도, 주목해야 할 부분이 있습니다. D-MPNN의 결과와 Fingerprint의 결과가 다르다는 점입니다. 무엇이 우월하다라고는 이야기하지 않겠지만, 분자를 어떻게 보는가에 따라서 다른 해석이 가능해진다는 것입니다. 그리고 그 해석에는 나름 합리적인 이유가 있습니다.
처음에 말한 대로, 구조는 다르지만, 비슷한 역할을 할 수 있는 작용기들이 있습니다. 이런 것들이 유사성이 높게 학습된다면, 뼈대 구조 기반의 유사성의 한계에서 벗어날 수 있을 것입니다. 단, 이런 것이 학습되기 위해선 데이터의 다양성이 필요합니다. 모든 active 분자가 동일한 작용기만을 가졌다면, 이들의 상호작용적 유사체가 과연 active분자로 분류될 수 있을까요? 경험이 많은 사람이라면, 그것을 유사체로 분류할 수 있지만, 제한된 데이터만을 사용하는 머신에겐 어려울 것입니다. 한 가지 방법은, 다양성 높은 데이터로 pre-training 하여 유사성들을 먼저 학습할 기회를 주는 것입니다. 다른 방법은, 유사체들을 미리 정리해서 묶어둔 피처를 추가로 도입하는 것입니다. 예를 들어 원자 피처에 수소결합 donor, acceptor, aromatic ring, hydrophobic 같은 정보를 많이 넣어줄 수 있습니다.
해석 가능성을 도입하기 위한 아이디어는 있지만, 연구할 시간이 별로 많지 않네요.
혹시 같이 연구하실 분 있으면 언제라도 연락 주시길 바랍니다.
저는 비영리기관에서 일하고 있고, 아이디어를 제공하는 지원 업무가 제 본업에 가깝습니다.
제가 속한 기관은 공동연구에 대해서 특허 등 영리적인 이익을 추구하진 않습니다.
그래도 논문을 쓰게 된다면 계속 함께 논의를 하고 저자로 넣어주거나, 최소한 감사의 글이라도 써주시길 바랍니다.
아무리 비영리기관이라 할지라도, 어떤 기여를 했다는 것이 기관의 실적인데, 실적을 평가할 결과가 남지 않으면 업무로 인정되지 않습니다.
'Drug > Computer-Aided Drug Discovery' 카테고리의 다른 글
| 분자 가상합성: moleculer building block assembly (0) | 2022.04.02 |
|---|---|
| pybel: openbabel python interface, 수소 추가, protonation state (0) | 2022.03.29 |
| 인공지능 신약개발에서 데이터 생산의 중요성 (0) | 2022.03.19 |
| Enrichment Factor (0) | 2022.03.13 |
| 머신러닝 기반 분자 활성 예측 정확도가 낮은 이유 (0) | 2022.03.02 |