관련글:
https://novelism.tistory.com/241
Marvin JS demo를 사용해서 분자 SMILES, SMARTS 얻기
이전에 SMILES와 SMARTS에 대해 포스팅을 했습니다. 직접 SMILES과 SMARTS를 만드는 건 좀 번거롭고 어렵기에 보통은 분자 그림을 변환 툴을 사용합니다. chemaxon의 marvin을 이용해서 SMILES와 SMARTS를 얻을
novelism.co.kr
SMILES (Simplified molecular-input line-entry system) 은 분자를 문자열로 표기하는 방법 중 하나입니다.
기초적인 문법은 위키피디아에 잘 설명되어있습니다.
https://en.wikipedia.org/wiki/Simplified_molecular-input_line-entry_system
Simplified molecular-input line-entry system - Wikipedia
From Wikipedia, the free encyclopedia Jump to navigation Jump to search Chemical species structure notation SMILES generation algorithm for Ciprofloxacin: break cycles, then write as branches off a main backbone The simplified molecular-input line-entry sy
en.wikipedia.org
SMARTS (SMILES arbitrary target specification) 는 SMILES이 확장된 형태로, 분자의 하위 구조 탐색을 위해 사용하는 규칙입니다. https://en.wikipedia.org/wiki/SMILES_arbitrary_target_specification
SMILES arbitrary target specification - Wikipedia
From Wikipedia, the free encyclopedia Jump to navigation Jump to search SMILES arbitrary target specification (SMARTS) is a language for specifying substructural patterns in molecules. The SMARTS line notation is expressive and allows extremely precise and
en.wikipedia.org
SMILES 가 특정 분자 하나를 지칭하기 위해 사용되는데 비해, SMARTS는 임의의 원자(*, wildcard)나, 임의의 조건을 만족하는 원자, 혹은 더 세부적인 규칙을 지원하여 분자의 탐색에 적합합니다.
예를 들자면, [R] 이라고 한다면, ring에 속한 원자를, [R0] 이라 한다면 ring에 속하지 않은 원자를, [R1] 이라면 ring1개에만 속한 원자를, [R2]라면 2개의 ring에 속한 원자를, [r5]라고 하면 ring size가 5인 ring에 속한 원자를 지칭합니다. [#7;H2] 의 경우는 하면, 원자번호 7인 질소 중에서 수소 2개가 결합된 것들을 지칭합니다. 이런 규칙들에 논리 연산을 적용하면(and, or 등) 상당히 복잡한 규칙을 표현할 수도 있습니다.
SMARTS의 자세한 규칙은 https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html 에서 볼 수 있습니다.
Daylight Theory: SMARTS - A Language for Describing Molecular Patterns
4. SMARTS - A Language for Describing Molecular Patterns Substructure searching, the process of finding a particular pattern (subgraph) in a molecule (graph), is one of the most important tasks for computers in chemistry. It is used in virtually every appl
www.daylight.com
SMILES나 SMARTS는 daylight, OpenEye, ChemAxon 뿐만 아니라, rdkit, obabel, 등 오픈소스 프로그램에서도 지원하고 있습니다. 하지만 서로 구현이 달라서 다른 결과가 나오는 경우도 있으니 주의해야 합니다. (아마도 문법이 다르다기보다는 그 문법을 구현한 프로그램적인 버그인 것 같습니다.)
그냥 컴퓨터로 읽으라고 할 수도 있겠지만, Cheminformatics를 한다면, SMILES와 SMARTS를 직접 어느 정도 다룰 수 있는 편이 좋습니다. 특히 SMARTS 같은 경우는, rdkit이나 ChemAxon의 Marvin Sketch 같은 프로그램에서 하부 구조를 이용해서 원하는 pattern을 만들어줄 수도 있긴 하지만, 그럴 경우 조건이 너무 느슨하거나 너무 엄격해서 원하지 않는 분자들이 탐색되는 경우도 있습니다. 일부 원자의 경우는 임의의 원자 중에서 선택하고 싶었을 수도 있고, 반대로 일부 원자는 이웃 결합의 수, 수소 원자의 수를 명확히 명시하고 싶을 수도 있습니다. 내가 원하는 조건이 있을 때, 거기에 부합하는 분자만을 탐색하기 위해선 컴퓨터에서 만들어준 SMARTS를 고치거나, 처음부터 새로 만들어야 합니다.
굳이 SMILES는 직접 만 들일이 별로 없지만, SMARTS가 SMILES의 확장이기에 SMILES도 어느 정도 알아두는 편이 좋습니다.
기본적으로 분자는 원자와 본드로 이루어집니다.
원자는 C, N, O, Cl처럼 원자 기호를 직접 넣어도 되고, [#7]처럼 대괄호 안에 원자 기호를 넣어도 됩니다.
bond는 -(single) =(double) #(triple) $(quadruple), : (aromatic) 이 있습니다. 그리고 공유결합이 없는 (이온 결합 같은) 경우는 . 으로 표기됩니다. 예시 ([Na+].[Cl-])
SMILES에서도 원자와 본드의 표기가 기본입니다.
단, 수소원자는 다른 원자와 동등하게 취급하는 것이 아니라, 어떤 원자에 귀속되는 것처럼 취급합니다.
수소원자와 단일 결합은 보통 생략됩니다.
CCC 라고 한다면, C-C-C, [CH3]-[CH2]-[CH3] 을 의미합니다. 괄호를 쓰면, 원자를 좀 더 명확히 규정해서 쓸 수 있습니다.
이웃 수소의 수, 전하량 등을 표기할 수 있습니다.
formal charge가 없을 때 C, N, O, 에서 원자의 결합수가 4,3,2가 아니라면, 부족한 만큼이 수소로 채워져 있다고 생각할 수 있습니다.
aromatic bond의 경우도 생략이 가능합니다. aromatic ring에 속하는 원소는 소문자로 표기하는데, c, n, o 이럴 경우 aromatic ring : 은 주로 생략됩니다.
aromatic 분자를 표기하는 법이 aromatic 표기와 Kekule 표기가 있는 것은 SMILES도 마찬가지입니다. 두 폼 모두 사용 가능합니다. (하지만 구현된 툴마다 결과가 달라질 수는 있습니다.)
특히 문법에 모호한 점이나 사소한 에러가 있는 경우, 프로그램이 스스로 교정을 하는데, 이 교정된 형태가 프로그램마다 다르거나, 교정에 실패하고 에러가 나기도 합니다.
SMILES 가 어렵게 느껴지는 이유는, 분자가 하나의 체인이 아니기 때문입니다. ring도 있고, 곁가지도 있습니다.
1차원 서열이 아닌데, 1차원 문자열로 표기하다 보니 어렵게 느껴집니다.
분자를 SMILES로 표기할 때는 먼저 main chain을 하나 잡습니다. 보통 제일 긴 체인을 잡습니다. (node와 node 사이의 distance가 최대가 되는 두 노드를 양 끝으로 합니다. 시작이고 누가 끝인지는 어떻게 정하는지는 잘 모르겠네요. 질소가 있다면 질소를 시작으로 정하던 것 같은데...), canonical SMILES라고 하는 표준 규칙이 있긴 한데, 이것도 프로그램 별로 다른 결과가 나올 때가 있습니다.
나머지는 곁사슬 (side chain) 취급합니다. 곁사슬은 메인 체인에()를 삽입해서 만듭니다.
ring은 곁사슬만으로는 표기가 안되고, 원래 자리로 돌아와야 하기 때문에, 결합이 필요한 위치에 숫자를 붙입니다.
벤젠의 경우 c1ccccc1 같은 식으로 표기합니다.
잘 모르면 생략 없는 표기를 보면 좋습니다.
single, double 본드로 ring 표기하는 경우를 보여드리겠습니다. 인덱스를 표기하기 위해서 원자에 괄호 안에 :0, :1, :6 의 숫자를 붙였습니다. 1과 6 사이만 =이고, 나머지는 - 입니다. 원자1과 원자6 사이의 bond type을 적을 때는 ring index 숫자 바로 앞에 적습니다. 숫자 뒤에 적으면 숫자 다음 원자와의 bond type이 됩니다.
그런데, ring index 숫자는 2곳에 적히죠? 양쪽 중 한 곳에 적어주면 되는데, 두 곳 다 적을 경우 rdkit에선 앞에 적은 것이 우선시되고, MarvinSketch에선 뒤에 적은 것이 우선시됩니다. 그러니까 두 위치 중 한곳에만 적는것이 좋습니다 아니면 두곳 다 같은 본드 타입을 적어야 합니다.
다음 예시의 분자들을 MarvinSketch로 그려보겠습니다.
'[CH1:1]=1[CH2:2]-C-C-C-[CH1:6]1' 그림1 좌측
'[CH1:1]1[CH2:2]-C-C-C-[CH1:6]=1' 그림1 좌측
'[CH1:1]-1-[CH2:2]-C-C-C-[CH1:6]=1' 그림1 좌측
'[CH2:1]=1-[CH2:2]-C-C-C-[CH2:6]-1' 그림1 중간
'[CH1:1]1=[CH1:2]-C-C-C-[CH2:6]-1' 그림1 우측

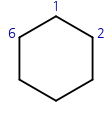

위키피디아에 있는 예시를 SMILES로 변환해봅시다.

먼저 C의 main chain만 적어보겠습니다.
NCCNC=CCNC=CCO
가 됩니다.
여기에 side chain 괄호를 삽입해봅시다. 가지의 깊이는 1입니다. (가지의 가지는 고려하지 않았다는 의미입니다.)
NCCN(CC)C(C=C)=CC(=CC=O)N(CCC)C=CC(=O)O
그다음은 가지의 깊이를 2로 올려보겠습니다. 괄호안에 괄호가 들어옵니다. 이 예제에서 해당사항은 C(F)=C 밖에 없습니다.
NCCN(CC)C(C(F)=C)=CC(=CC=O)N(CCC)C=CC(=O)O
그리고 ring 연결을 위해서 ring 인덱스를 삽입해줍니다.
N1CCN(CC1)C(C(F)=C2)=CC(=C2C4=O)N(C3CC3)C=C4C(=O)O
이러면 위와 같은 SMILES을 얻을 수 있습니다.
일일이 이렇게 손으로 만드는 건 시간도 오래 걸리고 실수할 가능성도 높아서 실제로 사용할 때는 MarvinSketch 같은 Cheminfomatics 툴킷에서 분자를 그린 다음에 SMILES로 저장해서 사용합니다.
MarvinSketch에선 분자를 그린 후에 마우스를 드래그해서 분자를 선택한 후, Edit -> Copy as Smiles를 선택하거나,
우클릭한 후 Copy As를 누르고 여기서 ChemAxon SMILES나 Daylight SMILES를 선택할 수 있습니다. |

웹에서 사용할 수 있는 프로그램들도 있습니다.
https://chemdrawdirect.perkinelmer.cloud/js/sample/index.html
ChemDraw JS Sample Page
chemdrawdirect.perkinelmer.cloud
분자를 그린 후에 Structure에서 Get SMILES 를 선택하면 됩니다.
SMARTS 사용법에 대한 글은 다음 글에 올리도록 하겠습니다.
'Drug > Computer-Aided Drug Discovery' 카테고리의 다른 글
| rdock: protein ligand docking 프로그램 (2) | 2021.10.05 |
|---|---|
| 분자 SMARTS 문법 및 rdkit에서 SMARTS 탐색 (0) | 2021.09.23 |
| 단백질 구조예측과 약물 결합 예측 (0) | 2021.09.16 |
| AutoDock Vina 1.2 가 나왔네요. (0) | 2021.08.13 |
| AlphaFold Protein Structure Database (0) | 2021.07.24 |